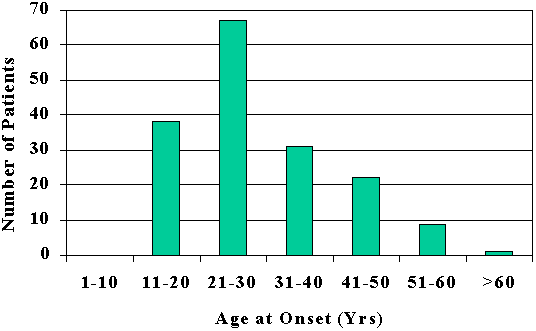|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
A Neurologist that I saw once gave me some information to take home from one of her medical books. To this day, it is still the BEST information that I have ever seen on Cluster Headaches...it's awesome! Even if you have suffered from Clusters your entire life and think you've read it all, you will find this info most interesting! It is quite a bit of reading, and quite difficult to follow at times, but well worth your time. Probably the easiest way to read it is if you print it out and read it when you get the chance.
Introduction
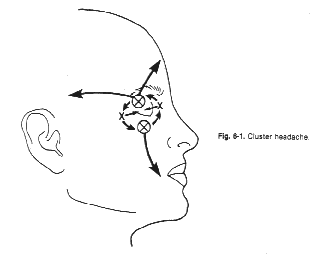
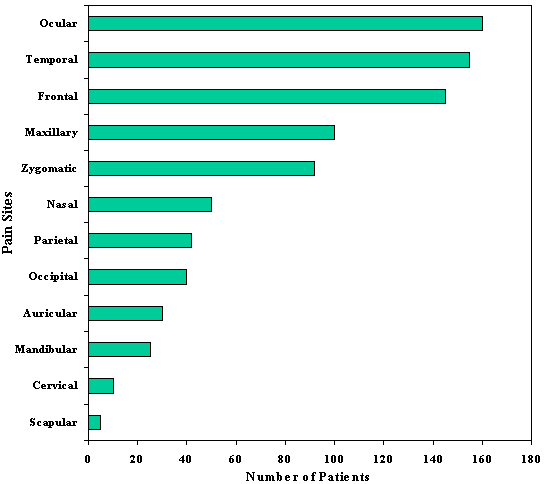
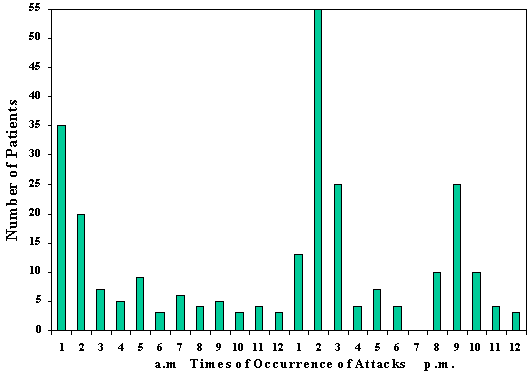
CLUSTER HEADACHEAlthough cluster headache ("migrainous neuralgia") had been recognized for over 100 years (von Möllendorff, 1867), Sir Charles Symonds' (1956) lucid account of this disorder brought it into focus. Recognition of the clinical entity was almost certainly retarded by a variety of confusing names that were given to this condition, such as erythroprosopalgia, Raeder's syndrome, spenopalatine neuralgia, ciliary neuralgia, vidian neuralgia, and histamine cephalalgia (Sjaastad, 1986; Grimson and Thompson, 1980). Cluster headache is now firmly established as a distinctive syndrome (Table 6-1) whose recognition is important, since it is likely to be responsive to treatment. The episodic type, the most common, is characterized by one to three short-lived attacks of periorbital pain (Fig. 6-1) per day over a 4 to 8 week period, followed by a pain-free interval that averages 1 year. The chronic form, sometimes called chronic migrainous neuralgia, which may begin de novo or several years after an episodic pattern has become established, is characterized by the absence of sustained periods of remission. Each type may transform into the other. The cluster syndrome is genetically, biochemically, and clinically different from migraine; propranolol is effective in treating migraine but has not been shown to be effective in cluster headache. Lithium is beneficial for the cluster headache syndrome and ineffectual in migraine. Nevertheless, the two disorders occasionally blend into one in occasional patients (Solomon, 1986), suggesting that their mechanisms bear some degree of commonality. CLINICAL FEATURESCluster headache has a prevalence of approximately 69 cases per 100,000 people, and is therefore far less common that migraine (D'Alessandro et al, 1986). Men are affected more commonly than women in a proportion of 6:1. Although most patients begin experiencing headache between the ages of 20 and 50 years (mean, 30 years), the syndrome may begin as early as the first decade and as late as the eighth decade (Fig. 6-2). Clearly, age alone is an insensitive diagnostic criterion (Krabbe, 1986). Women with cluster headache are more likely than men to begin experiencing attacks after the age of 50; among women, headaches usually do not correlate with menses, are likely to cease during pregnancy (Ekbom and Waldenlind, 1981), and may be initiated by use of oral contraception (Peatfield et al, 1982). Characteristics of PainThe pain of a cluster headache commences quickly, without warning, and reaches a crescendo within 2 to 15 minutes. It is often excruciating in intensity, and is deep, nonfluctuating, and explosive in quality; only occasionally is it pulsatile. In addition, 10 to 20 percent of patients report superimposed paroxysms of stabbing, icepick-like pains in the periorbital region that last for a few seconds and may occur once or several times in rapid succession; this paroxysmal pain usually heralds the end of an attack. The symptoms resolve in 1 to 2 minutes (Ekbom, 1975). The pain usually begins in, around, or above the eye or the temple (Fig. 6-3); occasionally the face, neck, ear, or hemicranium may be affected (Sutherland and Eadie, 1972). It is always unilateral, and generally affects the same side in subsequent bouts. However, it may shift to the corresponding region of the opposite side in 15 percent of patients (Manzoni et al, 1983b), usually for the duration of a bout, less often switching sides within a bout. Many patients prefer to be upright and active when an attack is in progress, but this is reported with a frequency that is not high enough to be useful diagnostically (Russell, 1981). Periodicity and Duration of the AttacksAttacks last from 30 minutes to 2 hours (mean of 45 minutes) in about 75 percent of cases. Occasionally, attacks - especially mild ones - may be as short as 10 minutes, whereas others may last as long as several hours. Attacks range in frequency from six per 24 hours to one per week, with a mean of one to two per day. Periodicity is a characteristic feature in about 85 percent of patients: attacks of pain tend to recur at the same hour each day for the duration of the cluster bout; many individuals also experience additional attacks that occur randomly throughout the day. About 75 percent of attacks occur between 9 p.m. and 10 a.m. (Russell, 1981). Manzoni et al, (1983b) found sharp peaks for attack frequency between 1 and 2 a.m., 1 and 3 p.m., and at 9 p.m (Fig. 6-4). Patients are awakened from sleep by pain paroxysms in about 50 percent of cases, usually within 2 hours of falling asleep (Lance and Anthony, 1971; Hornabrook, 1964). Nocturnal attacks are associated with rapid eye movement (REM) sleep about one-half the time in episodic cluster headache, but only rarely in the chronic form (Plaffenrath et al, 1986; Kayed and Sjaastad, 1985). Characteristics of the BoutsThe attacks of pain are clustered in cycles that usually last 4 to 8 weeks, and are followed by a pain-free remission in 90 percent of patients. On occasion, bouts may be as short as a few days or as long as 4 months; about 10 percent of those with established cluster tempos enter a chronic phase in which the attacks may persist for an average of 4 to 5 years (Ekbom, 1986). The later the onset of the episodic disorder, the greater the chance of it becoming chronic (Kudrow, 1980). Most patients experience one to two bouts per year; however, the interval between bouts ranges from 1 month to 2 years in 80 percent of the cases and between 6 months and 2 years in 60 percent (Kudrow, 1980). In rare instances it may be as long as 25 years (Hornabrook, 1964). Eventually, the bouts cease spontaneously, but more precise data on the natural history of the disorder are not yet available. Associated FeaturesRelated Symptoms and SignsLacrimation from the eye on the affected side is the most common associated symptom (Table 6-2). A blocked nasal passage, rhinorrhea, red eye, and sweating and pallor of the forehead and cheek are often found, but their absence does not exclude the diagnosis. These autonomic symptoms, although clinically apparent unilaterally, are present bilaterally, quantitaively more so on the symptomatic side (Saunte, 1984; Sjaastad et al, 1981). There is a rapid increase in heart rate at the onset of attacks and further rate variations become pronounced as the paroxysm proceeds, suggesting central autonomic regulatory instability. In general, a modest bradycardia occurs during an attack (Russell and Storstein, 1983). A transitory, partial Horner's syndrome (pupillary miosis and lid ptosis) occurs in two-thirds of patients when they are examined during attacks (Ekbom, 1970a) and is a useful sign in the differential diagnosis of facial pain. It is highly characteristic of the cluster headache syndrome and, after repeated occurrences, it may become a permanent feature (Riley and Moyer, 1971; Nieman and Hurwitz, 1961). The localization of the sympathetic lesion is of some interest. The ocular sympathetic innervation is a three-neuron pathway, comprising a first-order neuron: posterolateral hypothalamus to cord levels of C8 to T3 (the ciliospinal center of Budge); a second-order neuron: (preganglionic) ciliospinal center to super cervical ganglion; and a third-order neuron: (post-ganglionic) superior cervical ganglion to the pupillary dilator muscle, the eyelid muscles, and the facial sweat glands. It is believed by many observers that in cluster headache, involvement of the third-order neuron accounts for Horner's syndrome, which occurs as the result of distention of the wall of the internal carotid artery in the carotid canal, thus compressing the sympathetic plexus that invests the carotid wall. This argument is supported by three lines of evidence. The first is the observation that hyphidrosis is restricted to the forehead (Watson and Vijayan, 1982), which is consistent with a third-order neuron lision whereas more proximal lesions usually produce deficient sweating of the entire face (Morris et al, 1984). Second, supersensititivity of the miotic pupil to direct-acting sympathomimetic agents appears to place the lesion postganglionically (Fanciullacci et al, 1982; Vijayan and Watson, 1982); and third, angiographic changes of the carotid siphon during a cluster headache attack (Ekbom and Greitz, 1970) is also consistent with sympathetic plexus compression at this locus. However, others (Sjaastad, 1987) argue that there is sparse validation for the effects of conjunctival drugs on patients with central lesions (Malonety et al, 1980; Lepore, 1985). In fact, pupillary responsiveness and hyphidrosis patterns in patients with Horner's syndrome on a central basis are not very different from the results obtained with cluster headache patients (Van der Wiel and Van Gijn, 1986; Salvesen et al, 1987). Since the first-order hypothalamic neuron is uncrossed and, therefore, capable of generating ipsilateral symptoms, and for other reasons (see below) is an attractive patheogenetic locus, the issue, in my view, remains open. Focal neurologic symptoms of the type characteristic of migraine are very uncommon in patients with the cluster headache syndrome; however, occasional patients experience typical photopsia, teichopsia, facial paresthesia, or vertigo at the time of the attack. Precipitating FactorsSensitivity to alcohol during a cluster bout occurs in at least half the patients, and cases when the bout remits (Friedman and Mikropoulos, 1958); this alternating, on-off vulnerability is pathognomonic of the cluster headache syndrome. Patients who are sensitive to alcohol note that attacks are triggered within 5 to 45 minutes after the ingestion of modest amounts of alcohol: usually less than a single cocktail or glass of wine. The vast majority have noted that their sensitivity is less than total: alcohol triggers attacks in 70 to 80 percent of exposures. This factor, together with many patients' misinterpretations regarding inquiries into their drinking habits, may account for the low incidence of alcohol sensitivity in several reported series (Sutherland and Eadie, 1972; Symonds, 1956). A number of other precipitating factors have been noted in a smaller number of patients and include stress, relaxation, exposure to heat or cold, glare, hay fever attacks, and, occasionally, the ingestion of specific foods (chocolate, eggs, dairy products). There is some evidence that head trauma can precipitate the syndrome. Among the 180 patients studied by Manzoni et al (1983b), previous head injury was reported by 41, with loss of consciousness occurring in 20. This is significantly more frequent than is observed among patients with other types of headache; furthermore, in all patients in whom the head injury was lateralized and loss of consciousness ensued, the side involved corresponded to the side on which cluster headache later occurred. However, the mean latency in these cases was 9 years, which poses a serious question regarding the connection between head trauma and the cause of the cluster headache syndrome. Moreover, in an additional 11 of 15 patients who had undergone previous cranio-facial surgery, the side operated on was ipsilateral to that of the site of later-appearing cluster headache attacks. The latency between these latter events averaged 5 years. Kudrow has found no evidence that head trauma can precipitate the syndrome (Kudrow, 1980). Experimentally, attacks can be triggered in nearly all patients during a bout by the administration of 1 mg nitroglycerin sublingually (Ekbom, 1968), and in about 70 percent of patients by subcutaneous histamine (Horton, 1961). There is usually a latent period of 30 to 50 minutes before headache is triggered, whereas the peak peripheral and central vascular effects of nitroglycerin occur within 3 to 4 minutes of its administration and disappear in approximately 30 minutes (Bogaert, 1987). This, the appearance of headache does not coincide with the maximal circulatory effect of nitroglycerin, and the mechanism by which nitroglycerin causes headache remains unclear. A period refractory to pharmacologic provocation occurs after spontaneous or pharmacologically induced attacks and may persist for 2 hours or more (Ekbom, 1968; Horton, 1961). Therefore, valid provocative tests must be administered during an active bout, several hours after the attack has subsided. AMELIORATIVE FACTORSEkbom (1975) found that compression of the superficial temporal artery provided temporary relief for about 40 percent of his patients but just as often worsened the pain; carotid compression reduced the pain half the time, and worsened it in 25 percent of his cases. Vigorous physical exertion at the earliest sign of an attack can, in some patients, be remarkably effective in ameliorating or even aborting an attack (Atkinson, 1977; Ekbom and Lindahl, 1970). HEREDITARY DATAHereditary factors are significant in migraine and might be expected to be important in the cluster headache syndrome because of their mechanistic and pharmacologic similarities. However, it is uncommon to find other examples of cluster headache in the family history. Among Kudrow's (1980) 495 patients, 18 reported the presence of the syndrome in a parent. Migraine occurs no more frequently among the cluster headache population than among random population than among random populations (Andersson, 1985). When migraine predates the commencement of cluster attacks, migraine usually ceases when the cluster attacks begin (Bickerstaff, 1959); thus, although these disorders are biologically distinct, their mechanisms are probably connected. EXAMINATION FINDINGSA carefully elicited history is the key to diagnosis. There are no abnormalities to be found upon a physical or laboratory investigation other than Horner's syndrome occasionally. In approximately 70 percent of patients with cluster headaches, the carotid artery is palpably tender at several points in the neck (Raskin and Prusiner, 1977). The cluster headache syndrome, with all autonomic symptoms, on-off,alcohol sensitivity, ipsilateral tender carotid artery, and clocklike periodicity of attacks, has not been associated with any underlying intracranial structural abnormalities. There have been a few cases reported of lesions producing painful disorders resembling this syndrome, and some in which coincidental anatomic anomalies were demonstrated (Tfelt-Hansen et al, 1982; Mani and Deeter, 1982; Kurizky, 1984). Graham (1972) formed the impression that certain physical features seemed to be characteristic of cluster headache patients. These include a ruddy complexion, multifurrowed and thickened skin, and a broad, prominent chin: all contributing to a "leonine" facial appearance. However, these observations were uncontrolled, and I have been unable to confirm them. There is no evidence that important psychological variables bear on this syndrome (Cuypers et al, 1981; Kudrow, 1980). PATHOPHYSIOLOGYThe traditional designation of cluster headache as a vascular headache disorder is probably inappropriate; the vascular alterations that occur appear to be epiphenomenal, as they are in migraine, resulting from a primary CNS discharge. The hypothalamus may well be the site of such activation, containing posterior cells that regulate autonomic functions and anterior nuclei that serve as the major circadian pacemaker in mammals (Moore-Ede et al, 1983), both of which are necessary to explain the clinical symptoms and the uncanny periodicity of cluster headache attacks. The "biologic clock" is serotonergically modulated (Mason, 1986) and is connected anatomically to the eye (Sadun et al, 1984). The drugs effective in the treatment of the cluster headache syndrome enhance serotonergic neurotransmission, as also occurs in the treatment of migraine.` This suggests that unstable serotonergic neurotransmission, at different loci, may be common to both disorders. In this section, we will review, among other data, the evidence supporting the tantalizing speculation that the cluster headache syndrome may be the result of an antidromically discharging biologic pacemaker. Biologic ClocksA pacemaking mechanism in mammalian brain controls circadian rhythms (from the Latin circa diem, about 1 day), which are endogenous daily cycles. The most important of these biologic clocks is believed to be the suprachiasmatic nuclei (SCN): two small cell groups in the anterior hypothalamu7s just dorsal to the optic chiasm (Schwartz et al, 1987; Moore and Card, 1985; Turek, 1985). The pacemaker generates circadian rhythms, couples them with one another, and synchronizes them with external environmental events. Events in the internal milieu are arranged in temporal sequence to permit maximal adaptation to synchronized in phase and period, by time cues such as the daily light/dark cycle; a visual pathway from retina to the SCN is necessary to mediate entrainment (Sadun et al, 1984). The function of this system is to maintain daily order in physiologic processes, such as enzyme activities, body temperature, hormone secretion, and some behaviors. A disordered pacemaker may result in illness. For example, there is evidence that in jet lag and in manic-depressive illness, circadian rhythms may not be synchronized with one another or with the sleep-wake cycle (Wehr et al, 1983). Under normal conditions, a rhythm generated by the pacemaker is transmitted to synapses where a receptor rhythm evokes second messenger elaboration, which, in turn, modulates neurotransmission (Kafka et al, 1983). Lithium is believed to act on this second messenger system (see below). The SCN project to, and receive afferents from, the midbrain periaqueductal gray matter (Moore, 1983), so that a functional link to the pain-modulating system is feasible. Furthermore, serotonin-containing terminals arising from the midbrain dorsal raphe nuclei distribute in a dense plexus in the SCN and are capable of serotonin uptake. There is evidence that serotonergic mechanisms are involved in the expression rather than the generation of circadian rhythms. The SCN neurons are responsive to serotonin and its release via activation of the midbrain raphe projection to the SCN (Groos et al, 1983). Intrinsic pacemaker frequency appears to be modulated serotonergically by a mechanism that has not yet been established. It is interesting that lithium experts prominent effects upon circadian rhythms (Kripke and Wyborney, 1980; Kafka et al, 1982), possibly through an enhancement of serotonergic neurotransmission (Blier and De Montigny, 1985). Apart from the circadian periodicity of individual attacks and the periodic recurrence of bouts of cluster headache, further evidence of the role of a central pacemaker comes from hormonal studies among patients. Dampening of secretory circadian rhythms has been shown for melatonin, cortisol, testosterone, á-endorphin, á-lipotropin, and prolactin (Waldenlind et al, 1984; Chazot et al, 1984; Facchinetti et al, 1986; Nappi et al, 1985; Waldenlind and Gustafsson, 1987) during bouts; most of these rhythms revert to normal during remissions. Additional support for a CNS disturbance as the source of cluster attacks comes from studies of brain stem auditory-evoked potentials. Slowed conduction (increased I-V interpeak latencies) was shown ipsilateral to the painful side and became more pronounced during paroxysms of pain; lithium treatment appeared to shorten the latencies (Bussone et al, 1986). Clinically, the concurrence of cluster headache and trigeminal neuralgia, the cluster-tic syndrome, in which both disorders are ameliorated by microvascular decompression of the sensory root of the trigeminal nerve, also points to a centrally mediated pain mechanism. Cerebral Blood Flow AlterationsDilatation of extracerebral arteries appears to be common to both migraine and cluster headache (Sakai and Meyer, 1978); enhanced pulsation of the intraocular vascular bed occurs during cluster attacks but not during migraine attacks (Hrven et al, 1972; Hrven and Sjaastad, 1977), underlining the involvement of the internal carotid artery and its branches in the cluster headache syndrome. Evidence that part of the pain of cluster headache is derived from dilatation of intracranial branches of the internal carotid artery stems from the observation of Thomas and Butler (1946) that pain may be relieved in some patients by the intrathecal injection of saline, which increases the cerebrospinal fluid pressure to 700 mm H2O. The importance of vascular dilatation in cluster headache has been emphasized in the past because headaches may be precipitated during a bout by vasodilators such as alcohol, histamine (Horton, 1941, 1952; Hardebo et al, 1980), and nitroglycerin (Ekbom, 1968); however regional cerebral blood flow (CBF) studies in the modern era have shown only inconsistent alterations in flow during attacks, lending no support to the idea that vasodilatation is necessary to the pain mechanism (Nelson et al, 1980; Krabbe et al, 1984). Drummond and Lance (1984) and Drummond and Anthony (1985) showed that the increase in extracranial blood flow and increased temporal artery pulsations that attended individual attacks usually followed the onset of pain in affected areas, which led them to a primary neural discharge. Reduction of the severity of angina pectoris and limb claudication has been noted during some cluster bouts (Ekbom, 1970b), suggesting that, at least in some patients, an alteration of arterial tone outside the carotid circulation also occurs. Biochemical MechanismsA search for biochemical agents has been made on the presumption that the cluster headache syndrome may be mediated by a disorder of humoral control of blood vessels. The prominence of lacrimination, perspiration, and suffusion of the conjunctivae is consistent with an excessive cholinergic discharge. This reasoning led Kunkle (1959) to examine cerebrospinal fluid for acetylcholine-like activity, which he found in 4 of 14 patients at the time of headache; it was not found in 7 patients with classic migraine. SerotoninSerotonin alterations are more subtle in patients with cluster headache than in migraine. Medina et al (1979) found modest elevations of serotonin in whole blood during attacks of cluster headache, whereas platelet serotonin levels fall precipitously during migraine attacks. Waldenlind et al, (1985) found low whole blood serotonin levels among cluster patients both during an active bout and during remissions, comparable to levels found among migraine patients. Erythrocyte CholineErythrocyte choline concentrations are low in cluster headache patients (de Belleroche et al, 1984); this is an interesting observation because lithium administration greatly increases erythrocyte choline levels, an effect that persists for months. The depressed choline level is not confined to the acute attack; it is also present between bouts. de Belleroche et al (1986) took these data a step further and showed that erythrocyte membrane phosphatidylcholine/cholesterol ratios were increased in cluster headache patients, indicating a reduced turnover of phosphatidylcholine in the red cell membrane. It is not yet clear whether these intriguing findings are related to the mechanism of the disorder. HistamineThe possibility that histamine may be involved is supported by the reportedly higher incidence of duodenal ulceration in patients with cluster headache (Ekbom, 1970b) as well as by the precipitation of attacks with small amounts of this substance. Anthony and Lance (1971) and Medina et al, (1979) have shown that there is a modest increase in whole blood histamine during an attack; furthermore, elevations of urinary histamine were found in four of eight patients during cluster attacks (Sjaastad and Sjaastad, 1970). These reports are challenged by the lack of change in the catabolic pattern of intravenously administered C14 histamine in patients with cluster headache (Beall and Van Arsdel, 1960) and, since histamine is localized peripherally to basophilic leukocytes, caution is advised when interpreting whole blood levels (Porter and Mitchell, 1972). Patients with chronic myelogenous leukemia have very high blood levels of histamine but do not report headache. Furthermore, antihistaminic agents are disappointingly ineffective, as also has been histamine desensitization. It has been apparent for some time that there are at least two histamine receptors, since some of the effects of histamine are not blocked by the usual antihistaminic agents (Ash and Schild, 1966). Substantial evidence for two histamine-induced vasodilatation is only partly reversed by H1 antagonists (Hardebo et al, 1980), and it now appears likely that bot H1 and H2 receptors are present in the carotid vascular bed (Saxena, 1975), the availability of H2 antagonists has renewed the interest in testing the role of histamine in cluster headache. Anthony et al, (1978) and Russell (1979) have used H1 and H2 antagonists in the therapy of cluster headache, without clear success. It is possible that the elevation of blood histamine is the result of episodes of paroxysmal vascular instability, since histamine is but one of a group of diverse substances that includes the kinins, prostaglandins, and others that are released from tissues during injury or inflammatory reactions (Beaven, 1976). Mast CellsAppenzeller et al (1981) found that mast cells, the major repository of histamine in many tissues, are found in increased number in the skin of the painful temporal area in cluster headache patients; this effect is particularly striking within the first 10 hours after a cluster attack. Mast cell numbers in patients outside of a cluster period are similar to those of migraineurs (Appenzeller, 1987), which suggests that these cells are increased in both types of headache as a secondary event. No differences have been found in the dermal response to histamine among cluster headache patients when the painful side was compared to the opposite side (Bogucki and Prus¡nski, 1985), which lends no support to the idea that the periodic release of histamine might stimulate trigeminal nerve endings and thus be directly implicated in the mechanism of an attack. TREATMENTThe original therapy in this condition was based on administration of a vasoconstrictor such as ergotamine to antagonize the paroxysmal vasodilation that appeared to be the cardinal mechanism of pain production. However, the efficacy of agents that have no direct vasoactivity has cast doubt on the vasoconstrictor hypothesis. it appears more likely that the different treatment modes discussed in this section are successful because they stabilize serotonergic neurotransmission. The Acute AttackThe most satisfactory treatment is the administration of drugs to prevent cluster attacks until the bout is over. Several effective pharmacologic options may be chosen. However, whichever agent is used, there may be a lag period of days or, occasionally, weeks before effective suppression is achieved. For this reason, it is essential to attempt to treat the individual attacks until they can be prevented. Since the attacks are so brief and have such a rapid crescendo, orally administered, slowly absorbed drugs are generally ineffectual, but inhalational agents are often useful. ErgotamineThe ergotamine aerosol at a dosage of 0.36 to 1.08 mg (one to three inhalations) produces peak plasma levels of ergotamine within 5 minutes (Ekbom et al, 1983) and is effective about 80 percent of the time (Speed, 1960; Kudrow, 1980). Two important aspects of the use of the aerosol are often overlooked and account for may apparent treatment failures:
OxygenThe inhalation of 100 percent oxygen, via a tight-fitting mask at a flow rate of 8 to 10 liters/min for 10 to 15 minutes is dramatically effective for about 80 percent of those patients for whom this approach is feasible; oxygen is particularly effective for nocturnal attacks. This mode of therapy, although recommended by Horton (1952) over 35 years ago, has only recently been substantiated by controlled trials (Kudrow, 1981; Fogan, 1985). Oxygen inhalations may be repeated up to five times per day. Sakai and Meyer (1979) have shown that marked cerebral vasoconstriction results from the administration of 100 percent oxygen during cluster headache attacks. Whether this effect is direct or is mediated centrally has not been studied. Oxygen also stimulates the synthesis of serotonin in the central nervous system (CNS) (Costa and Meek, 1974). LidocaineKittrelle et al (1985) showed that the intranasal instillation of 1 ml 4 percent topical lidocaine was effective in terminating attacks in four of five patients. The patients were instructed to lie supine, with their heads extended backward 45 degrees and rotated 30 to 40 degrees toward the side of the headache. One ml lidocaine was slowly dropped into the nostril ipsilateral to the pain, and the patient's position was maintained for several minutes. If nasal congestion made it impossible to deliver the anesthetic solution to it target, the patient was initially treated with a few drops of intranasal 0.5 percent phenylephrine. These investigators believed that the lidocaine reached the sphenopalatine fossa and anesthetized the sphenopalatine ganglion. That is certainly possible but, and addition, local blockade of terminals of the trigeminal and glossopharyngeal nerves intranansally could also decrease afferent activity to the spinal trigeminal nucleus, stopping the pain because of convergence of the sensory elements of cranial nerves V, VII, IX, and X. The beneficial effect of topical lidocaine surely does not implicate the sphenopalatine ganglion in the mechanism of cluster headache. I find lidocaine to be an extremely useful therapeutic adjunct. The 1 ml dose may need to be repeated once or twice. For many patients, the application of lidocaine via the continuous pumping of a plastic nasal spray bottle is superior. In my experience, lidocaine is useful for about 60 percent of patients. DihydroergotamineFor the patient having an attack in the physician's presence, intravenous dihydroergotamine (DHE) rarely fails to about the episode within 5 minutes (Raskin, unpublished observations). MethoxyfluraneAnother option that is frequently useful is the inhalation of methoxyflurane, a rapid-acting analgesic. Patients are instructed to apply 10 to 15 drops rapidly to a handkerchief, pillow case, or paper tissue, form a funnel with their hands, and inhale for several seconds. They should be seated or reclining, since light-headedness may ensue. The analgesia produced by this agent lasts but a few minutes but, if administered early, the attack may be aborted (Raskin, unpublished observations). PhenylpropanolaminePhenylpropanolamine, taken orally, significantly shortens cluster attacks for many patients (Cohen, 1980). However, the rare occurrence of intracerebral hemorrhage following modest doses of this drug (Kase et al, 1987) makes it unsuitable in the face of viable safe alternatives. ProphylaxisThe major prophylactic drugs for the cluster syndrome are prednisone, lithium, methysergide, and ergotamine. Lithium appears to be particularly effective for the chronic form of the disorder. Pizotifen, nifedipine, verapamil, nimodipine, phenelzine, ergonovine, indomethacin, and cyproheptadine all have some documented efficacy. ErgotamineK. A. Ekbom (1947), Karl Ekbom's father, was the first to describe the use and effectiveness of ergotamine in preventing attacks; he used ergotamine tablets, 2 mg two to three times per day, and obtained good results in 13 of 16 patients. Two mg taken orally or 1 mg rectally 2 hours before an expected attack can be particularly effective, especially for nocturnal attacks. The advantage of ergotamine in this condition is that its clinical effect may be assessed within 24 hours, which is an obviously important consideration for patients experiencing several attacks of high-intensity pain every day. Long-term risk factors must be placed into the context of a relatively short exposure to whichever agent is chosen. It is interesting that ergotamine dependence is quite rare among cluster patients (Raskin, unpublished observations); once the bout is over, the drug is easily discontinued. I have encountered only two patients who did seem to generate additional cluster attacks during a bout through the use of large daily doses of ergotamine. The efficacy of ergotamine in treatment of the cluster headache syndrome has led many physicians to believe that here is more therapeutic overlap between this disorder and migraine than actually exists. Propranolol and amitriptyline, major drugs in migraine therapy, have no established role in the treatment of cluster headaches; conversely, lithium may exacerbate migraine (Peatfield and Rose, 1981). DihydroergotamineIntravenous DHE, given every 8 hours, has completely stopped cluster attacks in 20 consecutive patients. For 7 of them, the bout was over following 3 days of DHE treatment, but this could have been coincidental (Raskin, unpublished observations). PrednisoneFor the episodic form of cluster headache, prednisone is highly effective in over 75 percent of patients (Kudrow, 1978) (Table 6-3). The value of prednisone has been established in a double-blind study (Jammes, 1975), and it is clearly a first-line drug, if not the drug of choice in this situation. The dosage has varied from 10 to 80 mg daily in various studies (Couch and Ziegler, 1978). I use 80 mg per day for 7 days and then rapidly taper the dosage over 6 days. Pain paroxysms usually cease within hours for the first dose. If there is no response after 48 hours, prednisone should be stopped and an alternative therapy instituted. If, while the dosage is being tapered, headaches return, prednisone may need to be continued for the duration of the cycle, preferably on an every other day basis, at dosages less than 140 mg weekly. Many patients have to take prednisone for 5 or 6 days, but then stop it and find that the bout is over. This has happened often enough to convince me that prednisone can actually terminate the bout for about 20 percent of patients. Watson and Evans (1987) made similar observations in 2 of 11 steroid-treated patients with the chronic form of the disorder. It is, therefore, worth treating the chronic patients with a 2-week course of corticosteroid, with the aim of interrupting the cycle, although in most headaches return when the dosage is tapered. I have seen several patients who were unresponsive to full dosages of prednisone, but who responded spectacularly to triamcinolone at dosages of 32 mg per day. The remarkable efficacy of corticosteroids in this syndrome is not easily explained. McEwen et al, (1986) have reviewed the many potent actions of the corticosteroids on the CNS, but these are just beginning to be defined. De Kloet et al (1986) studied the relationship of corticosteroids to the serotonergic projection from the dorsal raphe nucleus to the hippocampus in rat brain; they found that corticosteroids exerted tonic control on serotonergic neurotransmission in this system. MethysergideMethysergide is effective for about 70 percent of patients with the episodic form of cluster headache; it is far less effective for the chronic form (Kudrow, 1980) (Table 6-3). The dosage range is 4 to 10 mg/day, with occasional patients requiring as much as 16 mg/day to stop the attacks. The major, rare hazard of retroperitoneal fibrosis, which may appear after several months of use of methysergide is not an important consideration when the drug is used to treat episodic cluster attacks, which usually requires only 4 to 6 weeks of drug exposure. Improvement usually begins within the first few days of therapy; occasionally responses may be delayed for 10 to 14 days. LithiumThe discovery by Ekbom (1974a,b) of the effectiveness of lithium for the prophylaxis of cluster headache has had a major impact on the treatment of this condition. Many studies have now been published regarding the use of this agent and have been reviewed by both Ekbom (1981, 1986) and Manzoni et al (1983a). The dosage has varied from 300 to 1500 mg daily, averaging 600 to 900 mg/day. Kudrow (1980) has found that about 10 percent of patients require only 300 mg/day. Favorable responses occur within 2 weeks of commencing treatment, usually within the first week. The chronic form of cluster is particularly well-suited for lithium treatment, with about 80 percent of patients substantially improved and remaining so for substantial periods of time (Table 6-4). During successful lithium therapy, about 60 percent of patients experience bursts of short cluster periods that are generally mild and of short duration. Ergotamine added to lithium at dosages of 2 to 4 mg/day is a highly effective strategy, even when either drug alone has been ineffective (Stagliano and Gallagher, 1983). In Kudrow's (1980) experience, approximately 40 percent of his patients treated with lithium require such concomitant ergotamine prophylaxis for complete relief of headache. In Ekbom's (1981) experience and in ours, lithium therapy blocks alcohol-provoked attacks in most patients with the chronic form of cluster. Following the cessation of lithium therapy, about 20 percent of patients experience long periods of remission, their disorder having transformed to the episodic form (Kudrow, 1980), which raises the possibility that lithium may alter the natural course of the disorder. The episodic form of cluster is less responsive to lithium treatment when it is begun after a bout has commenced. Three-fourths of the 68 patients studied by Manzoni et al (1983a) improved by more than 60 percent (Table 6-4). However, when used for subsequent cluster cycles, lithium was considerably less effective. These investigators made the interesting observation that four of their patients who experienced and average of more than four cluster bouts annually, when treated with lithium uninterruptedly for over 1 year, sustained no further attacks during lithium treatment. When lithium was withdrawn, attacks resumed within 1 to 3 weeks. Ekbom (1981) made the same observation regarding four similarly treated patients. Clinical Pharmacology. Lithium is readily absorbed from the gastrointestinal tract and peak plasma levels are reached 2 to 4 hours after its ingestion. It should be given in three divided doses, initially 900 mg daily, which may be decreased if side effects appear by breaking the tablet formulation of the drug into halves and adjusting the dosage downward. Gastrointestinal side effects may be considerably attenuated by using lithium citrate syrup, 300 mg/teaspoonful, which may be diluted in iced fruit juice. Lithium displaces intracellular sodium, and sodium depletion promotes its retention; the risk of intoxication is lessened by avoiding a low-salt diet and the use of natriuretic drugs (Reisgerg and Gershon, 1979). Plasma levels obtained 12 hours after the last dose should be maintained below 1.2 mEq/liter and should be measured weekly during the first few weeks of therapy, but only infrequently thereafter if the dosage is in the low range and the patient is free of side effects. Side Effects. The most common side effects are tremor, polyuria, nausea, diarrhea, and gait unsteadiness and are dose-related. High plasma levels may produce myoclonic jerks, dysarhria, hypotension, convulsions, and renal failure; rigidity of the limbs and fasciculations are characteristic features of moderate to severe degrees of intoxication (Sansone and Ziegler, 1985). Hemodialysis is the most effective means of dealing with serious acute toxicity. Fortunately, the relatively low dosage of lithium required to treat cluster headache successfully rarely gives rise to the more serious side effects. Lithium also suppresses thyroid function, occasionally resulting in hypothyroidism that may be irreversible (Medical Letter, 1980). The polyuria produced by lithium is mitigated by amiloride by blunting the inhibitory effect of lithium on water transport in the renal collecting tubule (Batlle et al, 1985). Irreversible renal concentrating defects occur rarely (Vestergard and Amdisen, 1981; Waller et al, 1984: De Paulo et al, 1986). Baseline studies of renal and thyroid states should be obtained before treatment is begun. Reversible intracranial hypertension has also been caused by lithium (Saul et al, 1985), so regular funduscopic examinations should be performed. There are a number of troublesome drug interactions. Lithium intoxication can occur with the concurrent use of methyldopa, tetacycline or indomethacin; there are, in addition, isolated reports of CNS toxicity with the concurrent use of haloperidol, thioridazine, carbamazepine, or phenytoin (Medical Letter, 1980). Mechanism of Action. Lithium has no effect on cerebral hemodynamics in patients with cluster headaches (Okayasu et al, 1984). On the other hand, there is substantial evidence that lithium stabilizes and enhances serotonergic neurotransmission in the CNS, particularly in the hippocampus, the site at which both S1 and S2 receptors are down-regulated during lithium treatment (Treiser et al, 1981; Blier and De Montigny, 1985; Hoffa et al, 1986). It is curious that serotonergic nerve fibers appear in human temple skin after lithium treatment (Dhital et al, 1985). As discussed above (see discussion of biologic clocks), the hypothalamic pacemaker is serotonergically innervated, and perhaps it is on this basis that lithium has prominent effects on circadian rhythms, acting to slow and alter them (Kafka et al, 1982; Kripke and Wyborney, 1980). The molecular site of action for lithium is currently believed to be at a second messenger system in the CNS (Worley et al, 1987). The polyphosphoinositides are a group of membrane phospholipids that, when activated, release two products both of which act as second messengers: Diacylglocerol and inositol triphosphate. This second messenger system is particularly abundant in brain and modulates many aspects of synaptic transmission. Lithium's actions on the phosphoinositide cycle may underlie its therapeutic actions (Drummond, 1987). That a biologic pacemaker is an important site of lithium's action is supported by the effectiveness of this agent in cyclical migraine and by a curious group of patients we have encountered over the past 10 years with "hypnic headaches." Six elderly patients, all but one men, between the ages of 65 and 77 began to be wakened from sleep regularly, at the same time each night, often during a dream, by a diffuse headache that lasted for 30 to 60 minutes, unassociated with any autonomic symptoms. In all cases, headache ceased after 300 to 600 mg of lithium was taken at bedtime (Raskin, unpublished observations). Pizotifen, CyproheptadineEkbom (1969) showed in a controlled trial with 28 patients, that 2 to 3 mg pizotifen daily produced excellent or good results in nearly 60 percent. Cyproheptadine, closely related to pizotifen pharmacologically, also has value in this disorder (K. Ekbom, personal communication), but the side effects of weight gain and sedation may be troublesome. IndomethacinChronic paroxysmal hemicrania is subgroup of the chronic form of cluster headache, is highly responsive to indomethacin. It is characterized by 15 or more focal attacks of head pain daily, each lasting for about 15 minutes. Mathew (1981) identified a transitional group of patients with the chronic form of cluster headache, who had attacks of headache of sudden onset, lasting 20 minutes, and occurring five times daily; four such patients were treated with indomethacin at 150 mg daily, with a crossover period to placebo, and clearly benefited. Similarly, Kudrow et al (1987) described six indomethacin-responsive patients with a cyclic form of paroxysmal hemicrania. Calcium Channel AntagonistsMeyer and Hardenberg (1983) and Mullaly and Livingstone (1984) have shown in small studies that verapamil, nifedipine, and nimodipine all reduce that frequency of attacks of the chronic type of cluster headache. I used nimodipine at doses up to 240 mg/day in 15 patients who were totally refractory to previous pharmacotherapy; 7 became headache-free for the 6 month duration of the open study. Nifedipine is quite effective in many patients at dosages that range between 40 and 120 mg daily. Occipital Nerve Steroid BlockadeAnthony (1985) studied 20 patients with cluster headache, 12 with the episodic and 8 with the chronic type. Blockade of the greater occipital nerve ipsilateral to the side of attacks was accomplished with lidocaine and 120 mg methyprednisolone acetate in polyethylene glycol. Attacks were arrested for 5 to 73 days; when lidocaine alone was used for the procedure, the attacks were not arrested, thus, supporting the importance of corticosteroid in the procedure. I have used steroid (betamethasone) block in 10 patients with the chronic disorder and produced remissions in 6 of them that lasted 7 to 21 days (Raskin, unpublished observations). I regard this as a useful temporary procedure for the desperate patient, to allow time for the next pharmacotherapeutic procedure to be implemented. The mechanism of this procedure is nonspecific; it involves decreasing afferent impulses to the spinal trigeminal nuclear complex. Invasive ProceduresFor patients who are completely refractory to all known medical therapy and continue to experience repeated attacks of pain chronically, a number of aggressive procedures have been implemented. These include percutaneous glycerol injections into the trigeminal cistern, trigeminal sensory rhizotomy, percutaneous radiofrequency trigeminal rhizotomy, superficial petrosal neurectomy, trigeminal branch avulsion, and decompression of the nervus intermedius (Ekbom et al, 1987; Watson et al, 1983; Onofrio and Campbell, 1986; Solomon and Apfelbaum 1986). The overall benefit from these procedures is about 50 percent, with no one procedure holding superiority. Since repeated injections of local anesthetic of the gasserian ganglion may provide long-lasting but temporary relief, I have used steroid injections of the maxillary division of the trigeminal nerve in four patients with infraorbital pain. Pain relief occurred in all four, lasting 10 to 34 days (Raskin, unpublished observations). Treating the PatientFor the patient in the throes of a first bout of cluster headache, corticosteroids and ergotamine hold the advantage that their beneficial effects can be assessed within 24 hours. A 10- day course of prednisone or triamcinolone treatment holds the promise of terminating the mechanism of the bout, and therefore, in my view, these are the drugs of choice. I also provide the patient with 4 percent topical lidocaine and inhalational ergotamine in the event that the first strategy fails. Should both ergotamine and the corticosteroids fail, methysergide, lithium, ergonovine, and phenelzine are the next therapeutic modes, to be used, in that order. Cigarette smoking should cease, at least for the duration of the bout, since in about 5 percent of patients this is a factor leading to drug refractoriness (Raskin, unpublished observations). For the chronic cluster headache patient, lithium, nifedipine, and nimodipine are the major choices. Some of these patients are responsive to indomethacin (Watson and Evans, 1987), and I have found several responsive to phenelzine. Occasionally, chlorpromazine has been useful at dosages in the 200 to 700 mg per day range (Caviness and O'Brien, 1980).
Table 6-1. Clinical
Stereotype of the Cluster Headache Syndrome
Fig. 6-3. Location of
maximal pain during cluster attacks in 180 patients.
Fig. 6-4. Times of
occurrence of cluster attacks in 180 patients.
Table 6-2. Associated Symptoms
in Cluster
Table 6-3. Treatment of
77 Episodic and 15 Chronic Cluster
Table 6-4. Results of
Lithium Treatment in the Episodic
|